
10 de Noviembre: Día mundial de la concientización del cáncer neuroendocrino:
Los tumores neuroendocrinos constituyen un grupo heterogéneo de tumores. Las localizaciones más frecuentes son el páncreas, el tracto digestivo y el pulmón, este tipo de neoplasias pueden surgir en prácticamente cualquier órgano del cuerpo, debido a que estas células se distribuyen en la etapa embrionaria por todo el organismo, a través de las crestas neurales, las glándulas endocrinas, los islotes y el sistema endocrino difuso.
Las células neuroendocrinas se caracterizan por producir hormonas, se almacenan dentro de las células y son secretadas a la sangre. Su función es ejercer un efecto de control sobre distintas células del organismo, de tal forma que cada hormona actúa sobre unos determinados tejidos de forma específica para mantener el correcto funcionamiento del organismo.
Los tumores neuroendocrinos son poco frecuentes, con una incidencia anual ajustada por edad de menos de 10 casos por 100.000 habitantes. Se ha observado un aumento en su incidencia en los últimos 30 años debido a varios motivos, principalmente a la mejoría de las técnicas diagnósticas y a la mejor identificación de los casos. Aunque su incidencia es baja, su prevalencia es significativa debido a la historia natural de la mayoría de estos tumores, de lento crecimiento y de larga supervivencia. Así, por ejemplo, los tumores neuroendocrinos suponen la segunda neoplasia avanzada más prevalente del tracto digestivo tras el cáncer colo-rectal.
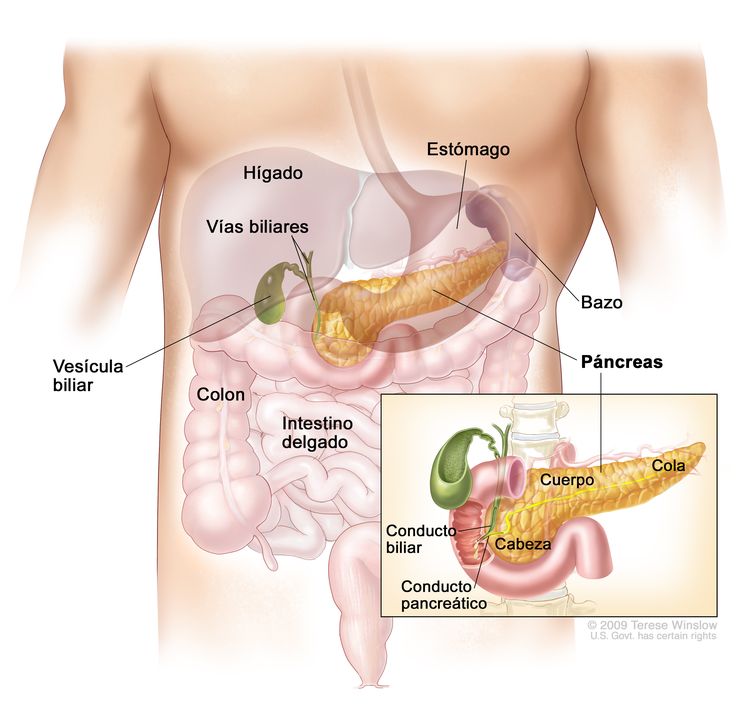
Causas y factores de riesgo
La mayoría de tumores neuroendocrinos son esporádicos y no presentan una causa ni unos factores de riesgo concretos conocidos. Sin embargo, en algunos casos pueden aparecer agregados en familias, dando lugar a síndromes hereditarios, cuando existen ciertas mutaciones germinales que pueden ser transmitidas en las sucesivas generaciones.
Entre los síndromes hereditarios destacan las neoplasias endocrinas múltiples, de herencia autosómica dominante, con potencial afectación de múltiples órganos.
Se distinguen dos formas de neoplasias endocrinas múltiples (MEN, por sus siglas en inglés).
- a) MEN 1
Es un síndrome hereditario caracterizado por el desarrollo de tumores endocrinos de páncreas (benignos o malignos) y duodeno junto con hiperplasia paratiroidea y adenomas hipofisarios. Para establecer el diagnóstico deben coexistir 2 de estas 3 lesiones. También se puede asociar a otras patologías como tumores carcinoides del pulmón y timo, tumores suprarrenales, lipomas (tumoraciones benignas de células grasas) y angiomas cutáneos.
Este síndrome se produce por la mutación germinal del gen MEN-1 situado en el cromosoma 11. Al heredarse de forma autosómica dominante cada hijo tiene una probabilidad del 50% de heredarlo.
Los síntomas clínicos son muy variados dependiendo de la localización tumoral y del tipo de hormona secretada, incluyendo alteraciones analíticas como hipercalcemia u otras como galactorrea, amenorrea, acromegalia o úlceras gástricas.
- b) MEN 2
Es muy infrecuente, con una incidencia menor a 1 de cada 1.000.000 habitantes. Se caracteriza por la presencia de carcinoma medular de tiroides (CMT). Se hereda también de forma autosómica dominante.
Existen tres formas, el MEN 2a, el MEN 2b y el carcinoma medular de tiroides familiar (CMTf). En el MEN 2a, el carcinoma medular de tiroides puede asociarse a feocromocitoma (un tipo de tumor maligno de la glándula suprarrenal) y a la hiperplasia de paratiroides.
En el MEN 2b, el carcinoma medular de tiroides se asocia a hábito marfanoide y neuromas. Puede asociarse también a feocromocitomas. Se han encontrado mutaciones del gen RET tanto en el MEN 2a como en el MEN2b.
Por último, las familias con miembros afectados por CMT como único signo de la enfermedad se engloban dentro del tercer subtipo, el CMTf.
Además de los síndromes MEN1 y MEN2, existen otros síndromes hereditarios poco frecuentes donde pueden aparecer tumores neuroendocrinos:
- Von Hippel Lindau: Se caracteriza por la predisposición al desarrollo de hemangioblastomas en retina y sistema nervioso central, feocromocitomas y/o paragangliomas, cáncer renal, quistes y tumores neuroendocrinos pancreáticos, entre otros. El gen responsable de la enfermedad, gen VHL, se localiza en el cromosoma 3.
- Síndrome feocromocitoma/paraganglioma familiar: Debido a mutaciones germinales en los genes SDH.
- Neurofibromatosis tipo 1: Síndrome hereditario caracterizado por la presencia de múltiples neurofibromas y manchas cutáneas características (manchas en café con leche), entre otras manifestaciones. Puede asociar neoplasias neuroendocrinas (feocromocitomas o tumores neuroendocrinos gastrointestinales) en aproximadamente un 2% de los casos.
Prevención y técnicas de diagnóstico precoz
No existen formas de prevenir las neoplasias neuroendocrinas esporádicas, dado que no existen factores de riesgo concretos. Tampoco se realizan exploraciones rutinarias para su diagnóstico precoz, teniendo en cuenta que además son tumores poco frecuentes. La concienciación social y la divulgación médica son las únicas medidas que se pueden implementar hoy en día para conseguir un diagnóstico más precoz de la enfermedad. Debido a la baja frecuencia, crecimiento lento, y presencia de síntomas inespecíficos, con frecuencia existe un retraso en el diagnóstico, que puede prolongarse incluso varios años. Por este motivo, es importante que el diagnóstico y tratamiento de los tumores neuroendocrinos se realice en unidades especializadas.
Clasificación
El grupo de neoplasias neuroendocrinas incluye dos grandes grupos que son los tumores neuroendocrinos (TNEs) y los carcinomas neuroendocrinos (CNEs). Con respecto a las localizaciones y subtipos tumorales, se incluyen los TNEs gastroenteropancreáticos (TNE-GEP), los TNEs pulmonares, los TNEs de origen desconocido, el feocromocitoma, el paraganglioma, el carcinoma medular de tiroides y el carcinoma de células de Merkel.
Aunque estas son las localizaciones más comunes, la ubicuidad de las células neuroendocrinas en el cuerpo hace que puedan aparecer neoplasias neuroendocrinas en otras localizaciones como el timo, el cérvix uterino, la vejiga, la próstata, etc.
Los TNE-GEP son los más frecuentes, localizados en el páncreas y el tracto digestivo. Se clasifican en función del índice Ki67 que refleja la agresividad tumoral. Actualmente se distinguen 4 grupos de agresividad creciente: los TNE-GEP Grado 1 (bien diferenciados, Ki67 <3%) son los que tienen un crecimiento más lento y mejor pronóstico, TNE-GEP Grado 2 (bien diferenciados, Ki67 3-20%), los NET-GEP Grado 3 (bien diferenciados, Ki67 >20%) y los carcinomas neuroendocrinos (pobremente diferenciados, Ki67 >20%), siendo estos últimos los de crecimiento más agresivo y peor pronóstico.
Otra forma de clasificación es en función de la existencia o no de un síndrome clínico producido por la liberación hormonal en algunos subtipos de estos tumores, de este modo se clasifican en tumores funcionantes o tumores no funcionantes.
Dentro de los funcionantes pancreáticos encontramos distintos tipos en función de la célula de los islotes pancreáticos de la que derivan y la hormona secretada, entre los que destacan el gastrinoma, insulinoma, glucagonoma, VIPoma y el somatostatinoma. Los TNE-GEP de origen intestinal también pueden ser funcionantes dando lugar a un síndrome carcinoide.
Subtipos y localizaciones
Dentro de los tumores neuroendocrinos gastroenteropancreáticos encontramos:
- Tumores neuroendocrinos bien diferenciados extrapancreáticos
Su localización más frecuente es el tracto gastrointestinal, seguida de la localización broncopulmonar.
- TNE gástricos: La mayoría son no funcionantes, sin metástasis. Aunque existe un subgrupo dentro de los mismos con un comportamiento más agresivo.
- TNE del intestino delgado: Son los tumores neuroendocrinos no pancreáticos más frecuentes, representando aproximadamente el 25%. Son más frecuentes en varones. En el momento del diagnóstico, más de la mitad de los casos presenta enfermedad no localizada. Se localizan más frecuentemente en el íleon distal y suelen ser multicéntricos. Son los que presentan con más frecuencia síndrome carcinoide.
- TNE apendiculares: Suelen ser diagnosticados de forma incidental tras procedimientos quirúrgicos por otros motivos (apendicitis, cirugías ginecológicas). Se caracterizan por tener un comportamiento poco agresivo.
- TNE de colon: La mayoría presentan metástasis al diagnóstico y suelen tener un comportamiento más agresivo.
- TNE de recto: Tienen menor tendencia a metastatizar que los colónicos. En algunos casos son localizados de forma precoz gracias a técnicas como la rectoscopia de rutina tras la presentación de clínica de heces con sangre, dolor o estreñimiento.
- TNE bronquiopulmonares: Dos tercios se encuentran localizados al diagnóstico.
- Tumores Pancreáticos Endocrinos
Se clasifican en función de la célula endocrina pancreática de la que derivan y el tipo de hormona que secretan:
- Gastrinoma: Son tumores funcionalmente activos que origina un síndrome de secreción inadecuada de gastrina llamado síndrome de Zollinger-Ellison. Un 20% son familiares, formando parte de MEN1.
Se localizan habitualmente en páncreas o en duodeno (principalmente en la primera y segunda porción duodenal). Algunos presentan metástasis en el momento del diagnóstico.
- Insulinoma: Son tumores habitualmente benignos, que producen insulina, lo que condiciona la aparición de hipoglucemia. Sólo una décima parte son malignos.
Un 5% se asocian con el síndrome MEN1.
- Glucagonoma: Es un tumor funcionalmente activo que secreta glucagón.
Un porcentaje elevado de los glucagonomas presentan metástasis en el momento del diagnóstico.
- Somatostatinoma: Tumor muy infrecuente habitualmente funcionante, secretor de somatostatina, de comportamiento maligno. Se localiza con la misma frecuencia en páncreas que en intestino delgado. Puede aparecer asociado con los síndromes neurofibromatosis tipo 1, MEN 1 y Von Hippel-Lindau.
- VIPoma: Tumores funcionalmente activos y con frecuencia malignos, que segregan polipéptido intestinal vasoactivo (VIP, por sus siglas en inglés) originando un síndrome de diarrea acuosa característico (Síndrome de Verner-Morrison). Suelen ser tumores solitarios, de gran tamaño que se localizan en la cola del páncreas.
La mitad son malignos, con metástasis en el momento del diagnóstico.
- Tumores endocrinos pancreáticos no funcionantes: Se caracterizan por no producir ningún síndrome derivado de la hiperproducción hormonal.
La localización más habitual es la cabeza de páncreas, y originan síntomas derivados del crecimiento local, como dolor e ictericia (coloración amarillenta de la piel y mucosas por ascenso de la bilirrubina en sangre). Son relativamente agresivos, y en más de la mitad de los casos presentan comportamiento maligno, con invasión local o metástasis. El análisis de mutaciones en el DNA de estos tumores mediante secuenciación masiva ha demostrado la presencia de alteraciones en MEN-1, ATRX/DAXX y mTOR. Diferentes fármacos se están ensayando para frenar estas vías de proliferación tumoral.
Otros tumores neuroendocrinos:
- Carcinomas pobremente diferenciados
Tienen un comportamiento más agresivo, suelen tener metástasis en el momento del diagnóstico.
- Carcinoma medular de tiroides
Representa entre el 3-5% de los carcinomas de tiroides. El 80% son esporádicos y el 20% hereditarios. En la mayoría de los pacientes la enfermedad se encuentra diseminada en el momento del diagnóstico. Secreta calcitonina entre otras hormonas, responsables de síntomas clínicos como la diarrea y flushing faciales.
- Carcinomas de la glándula suprarrenal
Es un tumor muy infrecuente. En algunos casos puede presentarse dentro de síndromes hereditarios como el Síndrome de Li-Fraumeni, entre otros.
- Feocromocitomas y paragangliomas
Los feocromocitomas derivan también de la glándula suprarrenal y pueden secretar unas hormonas llamadas catecolaminas. Entre un 15-20% aparecen fuera de la glándula suprarrenal recibiendo entonces el nombre de paragangliomas. Pueden ser esporádicos o englobarse dentro de síndromes hereditarios.
Signos y síntomas al diagnóstico
Tumores neuroendocrinos gastroenteropancreáticos
- Tumores neuroendocrinos no pancreáticos: Muchos TNE son encontrados de forma casual durante la cirugía por otros motivos, como apendicetomía o pancreatitis aguda. En caso de haber síntomas, éstos suelen ser inespecíficos o imprecisos como disconfort abdominal u oclusiones intestinales intermitentes, causando relativamente largos retrasos en el diagnóstico. Los TNE gástricos, duodenales y rectales suelen encontrarse de forma casual mediante endoscopia.
- Síndrome carcinoide: El síndrome carcinoide ocurre en algunos pacientes con TNE intestinales, y es debido a la producción hormonal, principalmente de serotonina.
Salvo algunas excepciones, sólo se manifiesta en presencia de metástasis hepáticas o, en ocasiones, pulmonares. Sin embargo, no todos los pacientes con metástasis hepáticas lo presentan.
El síndrome carcinoide se divide en típico y atípico. El 95% de los casos son típicos, siendo la sintomatología más presente el flushing o enrojecimiento cutáneo de la parte superior del cuerpo, diarrea, fibrosis cardiaca, sibilancias (silbido al respirar), disnea (dificultad respiratoria), telangiectasias (arañas vasculares) faciales y pelagra. El síndrome atípico (5%) consiste en flushing prolongado, cefalea, lagrimeo y broncoconstricción (estrechamiento de los bronquios). Ambos síndromes están desencadenados por mediadores hormonales, cuando estos alcanzan el torrente circulatorio. En el síndrome típico, el mediador principal es la serotonina.
Su tratamiento se basa en el uso de análogos de la somatostatina.
Tumores neuroendocrinos pancreáticos
Excepto en los Vipomas y los tumores no funcionantes, los primeros síntomas son debidos a una secreción hormonal excesiva, mientras que los síntomas tardíos son debidos a una diseminación del tumor (dolor, pérdida de peso o incluso sangrados).
– Insulinoma
Las manifestaciones clínicas son debidas a la hipoglucemia (bajada de azúcar) secundaria a la excesiva y no regulable producción de insulina.
Muchos pacientes tienen síntomas debidos a la falta de glucosa en el cerebro, como cefalea (dolor de cabeza), confusión, alteraciones visuales o carácter irritable. También pueden presentarse síntomas como sudoración, palpitaciones o temblores.
La clínica aparece típicamente en situaciones de ayuno o después de la realización de ejercicio.
El diagnóstico puede requerir de la medición conjunta de los niveles sanguíneos de insulina y glucosa realizada en el hospital.
– VIPoma
La sintomatología es debida al exceso de secreción de péptido intestinal vasoactivo (VIP, por sus siglas en inglés), y se caracteriza por la presencia de diarreas acuosas profusas, hipopotasemia (descenso del potasio en sangre), hipoclorhidria (descenso del cloro) y deshidratación. Este cuadro clínico también recibe el nombre de síndrome de Verner-Morrison. Su diagnóstico requiere la demostración de la elevación en plasma de VIP y la presencia de diarrea secretora (líquida) mayor de 700 ml/día.
– Glucagonoma
La sintomatología se debe a la excesiva secreción de glucagón y consiste en una dermatitis característica llamada eritema necrolítico migratorio, hipoaminoacidemia (descenso de aminoácidos en plasma), intolerancia a la glucosa e incluso diabetes mellitus, pérdida de peso y anemia.
El eritema necrolítico migratorio está presente en la mayoría de los casos y se inicia alrededor de la boca de forma anular y posteriormente en inglés, periné, nalgas y muslos y se extiende lateralmente produciendo ampollas y erosiones.
El diagnóstico se basa en la demostración de niveles elevados de glucagón en sangre.
– Somatostatinoma
Los somatostatinomas se caracterizan por la secreción de somatostatina, que es una sustancia capaz de inhibir la liberación de casi todas las hormonas intestinales y la responsable de la sintomatología clínica, que es poco específica y consiste en diabetes mellitus, litiasis (cálculos) biliar, diarrea, pérdida de peso, esteatorrea (diarrea con abundante grasa) e hipoclorhidria. Dado que estos síntomas son inespecíficos, en muchas ocasiones se trata de un hallazgo quirúrgico.
– Gastrinoma
Se caracteriza por la hipersecreción de gastrina, produciendo una estimulación de la secreción gástrica, con un aumento en la tendencia de formación de úlceras y diarrea.
Estas úlceras acostumbran a ser recurrentes y resistentes a los tratamientos convencionales. Está clínica también es conocida como síndrome de Zollinger-Ellison.
El diagnóstico se basa en la demostración de hipersecreción ácida gástrica y niveles elevados de gastrina no justificables por otras causas.
– ViPomas y tumores no funcionantes
Los ViPomas son capaces de secretar polipéptido pancreático, aunque no producen síndromes hormonales específicos reconocidos. Suelen diagnosticarse de forma tardía al producir metástasis a otros órganos.
Feocromocitomas y paragangliomas
Las manifestaciones clínicas suelen ser debidas a la secreción de catecolaminas: hipertensión, taquicardia, palidez, cefalea, sudoración, sensación de angustia, pérdida de peso. Pueden aparecer problemas cardiovasculares y accidentes cerebrovasculares. Debido a que estos síntomas son inespecíficos suele haber un retraso diagnóstico de unos 3 años desde el inicio de estos.
Estudios diagnósticos
Ante la sospecha de un tumor neuroendocrino se debe realizar:
- Analítica general – Estudio hormonal inicial en plasma y/u orina: La hormona estudiada dependerá del tumor neuroendocrino que se sospeche clínicamente. En los TEGEP debe realizarse la determinación de Cromogranina A sérica en ayunas y en los casos en que se sospecha el diagnóstico de un tumor carcinoide clásico un examen en orina de 24 horas de 5-HIAA (ácido 5-hidroxiindolacético).
- Pruebas de imagen. Medicina nuclear: Las pruebas de medicina nuclear tienen especial utilidad en el diagnóstico de tumores neuroendocrinos. Las más habituales son la gammagrafía con octreotido, el PET-Galio68 y el PET-FDG (glucosa). El objetivo es detectar la expresión de receptores de somatostatina (gammagrafía, PET-Galio68) o la actividad tumoral a través del consumo de glucosa (PET-FDG). De esta manera se puede determinar la agresividad del tumor y la sensibilidad a tratamientos dirigidos a los receptores de la somatostatina. La prueba con Galio68 es la más sensible actualmente para la evaluación funcional de TNE bien diferenciados.
- Gammagrafía con MIBG 131: Es la técnica más sensible y específica para el diagnóstico de feocromocitomas y paragangliomas.
- TAC de alta definición: Puede ser útil como prueba principal en los casos con gammagrafías negativas, y como estudio complementario en los casos con gammagrafías positivas al proporcionar imágenes mucho más precisas anatómicamente. El TAC es esencial para el seguimiento de la enfermedad metastásica y para seguir la respuesta a los tratamientos. Debido a las características especiales de los TNE-GEP el TAC debe ser trifásico para obtener las mejores imágenes posibles.
- Resonancia magnética nuclear (RMN): Presenta una gran sensibilidad para la detección de metástasis hepáticas. También es muy útil en la detección de otros tumores neuroendocrinos como el de tiroides o los de hipófisis.
- Estudio anatomopatológico: Debe realizarse una biopsia tumoral en todos los casos. Un estudio anatomopatológico reglado es esencial para realizar un correcto diagnóstico del tumor y una correcta clasificación.
Tratamiento
Las decisiones terapéuticas de los pacientes con neoplasias neuroendocrinas deben tomarse en el contexto de comités multidisciplinares que incluyan todas las especialidades involucradas en el manejo de esta enfermedad (cirugía, radiología, endocrinología, digestología, oncología, medicina nuclear, anatomía patológica). Además, es muy importante que el centro cuente con otras especialidades complementarias como puede ser nutrición, psico-oncología, enfermería oncológica, etc.
Además de la evaluación por un equipo médico, es fundamental conocer los recursos para pacientes disponibles en el entorno más cercano, como pueden ser las asociaciones de pacientes y los grupos de apoyo. En el ámbito de los tumores neuroendocrinos, la asociación de mayor presencia a nivel nacional es NET-España.
- Tumores neuroendocrinos gastroenteropancreáticos
- Enfermedad localizada
Ante una enfermedad localizada, el tratamiento de elección es la cirugía o la exéresis tumoral por técnicas endoscópicas en situaciones concretas, puesto que son las únicas modalidades de tratamiento que pueden lograr la curación.
- Enfermedad diseminada únicamente al hígado y tumor primario resecable
El hígado es el sitio de diseminación más frecuente de las neoplasias neuroendocrinas. Los TNE-GEP con diseminación únicamente hepática son subsidiarios de tratamientos radicales con cirugía completa, citorreductora o ayudada de técnicas de ablación local (radiofrecuencia).
Únicamente son candidatos a una resección quirúrgica completa entre el 10-20% de los pacientes con metástasis hepáticas. La realización de resecciones que supongan una extirpación de al menos el 90% del volumen tumoral (cirugía citorreductora) supone también un beneficio tanto clínico como en supervivencia, en particular en el caso de los tumores funcionantes.
Existen otras técnicas en el tratamiento de las metástasis hepáticas que tienen como objetivo disminuir la masa tumoral ya sea para intentar posteriormente una cirugía o bien paliar los síntomas derivados de la enfermedad. Estas son la embolización o quimioembolización.
- Enfermedad diseminada o tumor localmente avanzado irresecable
En la enfermedad diseminada a múltiples órganos, tanto la cirugía como los tratamientos ablativos locales antes comentados tienen un destacado papel en prevenir complicaciones, mejorar síntomas y aumentar la supervivencia.
Además, el abanico terapéutico es cada vez más amplio con tratamientos biológicos, quimioterapia, radionúclidos y fármacos de nueva generación.
- Análogos de somatostatina
Más del 80% de los TNEGEP expresan receptores de somatostatina. Por ello, se han utilizado análogos de la somatostatina con fines terapéuticos.
El tratamiento con análogos de somatostatina tiene un doble papel. Por un lado, controla el crecimiento tumoral, generalmente manteniendo estable el tamaño de las lesiones, y por otro controla la producción hormonal responsable del síndrome carcinoide y los síndromes funcionantes pancreáticos.
Actualmente disponemos de dos fármacos en este grupo de los análogos de la somatostatina, Octreotida LAR y Lanreotida Autogel. Ambos deben ser prescritos por un profesional con experiencia en el tratamiento de los TNEs y pueden ser administrados por profesionales de enfermería o por el propio paciente tras un adecuado aprendizaje. Debido a su liberación progresiva, únicamente es necesaria una dosis cada 28 días de manera intramuscular (octreotida) o subcutánea profunda (lanreotida). Adicionalmente también existen formulaciones de acción inmediata para uso hospitalario y control de síntomas refractarios.
El tratamiento con análogos de la somatostatina tiene una doble indicación: el tratamiento del síndrome hormonal secundario al TNE, como puede ser el síndrome carcinoide, o los síndromes asociados a los TNE pancreáticos funcionantes, como el VIPoma, el somatostatinoma o el glucagonoma; y el tratamiento antitumoral.
- Radionúclidos
El tratamiento con radionúclidos es una de las novedades de los últimos años en el tratamiento de los tumores neuroendocrinos. Desde 2017 disponemos de evidencia científica suficiente para utilizar en nuestro país el tratamiento con Lutecio radiactivo (177Lu-DOTATATE), especialmente en tumores neuroendocrinos de origen intestinal.
El tratamiento se basa en la administración de 177Lu-DOTATATE intravenoso en 4 dosis que se pautan cada 2 meses, en pacientes que hayan recibido previamente análogos de somatostatina y que tengan positividad en las pruebas funcionales (octreoscan o PET-Galio68).
La implementación de esta terapia en España se está realizando de manera progresiva debido a las dificultades logísticas que supone este tratamiento, aunque ya se encuentra disponible en los centros de referencia. Precisa una coordinación estrecha entre medicina nuclear, oncología, endocrinología y todos los servicios implicados para su correcta administración, evaluación y seguimiento.
- Quimioterapia
La sensibilidad de los tumores neuroendocrinos a la quimioterapia varía según su grado de diferenciación celular y la localización del tumor primario.
Los tumores de intestino delgado (carcinoides clásicos) muestran tasas de respuesta muy bajas a la quimioterapia. De hecho, la quimioterapia no se considera una opción de manera rutinaria en este tipo de tumores.
Los tumores neuroendocrinos del páncreas han sido clásicamente definidos como tumores más sensibles a la quimioterapia, con tasas de respuestas superiores, que en las series más recientes oscilan entre el 15-25%. Actualmente los esquemas de quimioterapia más ampliamente utilizados son los dobletes de estreptozocina con 5-fluoracilo o la temozolamida en combinación con capecitabina.
Los carcinomas neuroendocrinos pobremente diferenciados, muestran una alta tasa de respuestas a la quimioterapia, siendo este tratamiento la base de la terapia. Las combinaciones más habituales incluyen los platinos (cisplatino, carboplatino) en combinación con etopósido.
- Terapias dirigidas
Los avances de los últimos años en el conocimiento de la biología molecular de los tumores neuroendocrinos han dado lugar al desarrollo de múltiples estudios evaluando la eficacia y tolerabilidad de distintos grupos de fármacos dirigidos contra dianas moleculares.
Los fármacos inhibidores de receptores con actividad tirosina cinasa y efecto antiangiogénico (bloqueo de los vasos sanguíneos tumorales), así como los inhibidores de la proteína mTOR han logrado demostrar su actividad en TNE de origen pancreático. Es frecuente encontrar estos fármacos bajo la denominación de Inhibidores Tirosina Kinasa (TKI) o Inhibidores Multikinasa (MKI).
Sunitinib, un fármaco con actividad antiangiogénica, es el primero de estos nuevos fármacos que ha demostrado su eficacia en un ensayo fase III en TNE de origen pancreáticos y ha permitido su aprobación y utilización en esta población de pacientes.
Everolimus, un inhibidor de la ruta metabólica mTOR, ha demostrado eficacia en varios ensayos fase III en el contexto de los tumores neuroendocrinos pancreáticos, intestinales y pulmonares.
- Otros tumores neuroendocrinos
En la mayoría de los casos el tratamiento de elección cuando la enfermedad está localizada es la cirugía, en algunas ocasiones acompañada de radioterapia. En aquellos casos con enfermedad diseminada o recurrente el abanico de tratamientos sistémicos indicados puede ser amplio englobando la quimioterapia, radioterapia, análogos de la somatostatina y fármacos de diana molecular, en función del tipo de tumor.
Es importante destacar también la importancia del uso de medicación sustitutiva en aquellos casos en los que se haya extirpado una determinada glándula endocrina y el uso de tratamiento médico para el control del exceso de secreción hormonal cuando ésta se produce.
- Ensayos clínicos
En los últimos años han aparecido nuevos tratamientos y estrategias terapéuticas para el manejo de los pacientes con tumores neuroendocrinos. Estos tratamientos incluyen el desarrollo de nuevos fármacos biológicos, radionúclidos y la inmunoterapia.
El acceso a estos nuevos fármacos se restringe al ámbito de los ensayos clínicos y deben valorarse siempre en función de las preferencias de cada paciente y la disponibilidad del centro. Siempre es recomendable la derivación de pacientes con tumores neuroendocrinos a centros de referencia que dispongan de ensayos clínicos y experiencia en el manejo de esta patología.
Seguimiento
Los pacientes diagnosticados de una neoplasia neuroendocrina deben realizar revisiones periódicas durante años. La frecuencia de las revisiones puede ser variable en función del tipo de tumor, su extensión y la presencia o no de síntomas. En los pacientes con metástasis, el seguimiento suele realizarse cada 3 meses. En aquellos pacientes operados y libres de enfermedad, el seguimiento suele espaciarse y realizarse cada 3, 6 o 12 meses según la situación.
En muchos casos el seguimiento debe ser realizado por varios especialistas, siendo oncología, endocrinología y cirugía las más habituales.
En las revisiones, además de la recogida de síntomas y de la exploración física, suelen realizarse de forma periódica analíticas de sangre y/o orina, y determinadas pruebas de imagen en función del tumor (tanto de radiología como de medicina nuclear).
Dr. Niewiadomski Dario
MN: 110.535
Médico oncólogo